

Background: Gene Fusion events are common occurrences in malignancies, and are frequently drivers of malignancy. FISH and qPCR are two methods often used for identifying highly prevalent gene fusions/translocations. However, these are single target assays, requiring a lot of effort and sample if multiple assays are needed for multiple targets like sarcoma. High-throughput parallel (NextGen) DNA and RNA sequencing are also in current use to detect and characterize gene fusions. RNA sequencing (RNAseq) has the advantage that multiple markers can be targeted at one time and RNA fusions are readily identified from their product transcripts. While many fusion calling algorithms exist for use on RNAseq data, sensitive fusion callers, needed for samples of low tumor content, often present high false positive rates. Further, there currently is no single variable or element in NGS data that can be used to filter out false positive calls by extant callers. Individual sensitive fusion callers may be considered weak predictors of gene fusions. Combining their results into a single fusion call involves evaluating many elements, which can be a time consuming and difficult manual task. In order to achieve higher accuracy in fusion calls than can be achieved using individual fusion callers, we have combined the results of multiple fusion callers by use of an ensemble learning approach based on random forest models. Our method selects the best group of callers from among several callers, and provides an algorithmic means of combining their results, presenting a metric that can be immediately interpreted as the probability that a called fusion is a true fusion call.
Methods: Random forest models were generated with the randomForest package in R, and then tuned using the R caret package. Training data sets consisted of fusion calls deemed true by review and by orthogonal methods including PCR/Sanger sequencing and the commercial Archer™ fusion calling system. We present the results of training on calls made by five fusion callers Arriba, STAR-Fusion, FusionCatcher, deFuse, and Kallisto/pizzly. Logistic training variables (seen vs not seen by the fusion caller) were used for the five callers. Variables also included metrics for the magnitude and balance of coverage on either side of candidate fusion breakpoints reported by Arriba and STAR Fusion ("coverage balance") and a single metric consisting of the number of sequencing reads that cross the candidate breakpoint. The model was validated by 10-fold cross-validation on 598 fusion calls by the five callers.
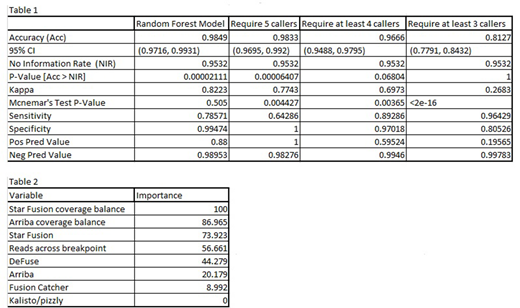
Results: The resulting model is superior to the simple strategy of requiring agreement by n of five callers, particularly with regard to specificity (Table 1). Also, "importance of variables," reported by randomForest, gauges the relative contribution of variables in the model. Here it shows that one caller, Kallisto\pizzly, does not contribute to the model (Table 2).
Conclusion: Random Forest modeling provides a viable means of combining gene fusion call data from multiple callers into a single fusion calling tool with improved performance over simple combinations of fusion calls. An additional benefit is seen in that building and evaluating such models can guide the selection of fusion callers, thereby eliminating non-contributory calling methods and ensuring optimal utilization of computational resources.
Thomas:NeoGenomics,Inc.: Current Employment. Mou:NeoGenomics: Current Employment. Keeler:NeoGenomics: Current Employment. Magnan:NeoGenomics: Current Employment. Funari:NeoGenomics: Current Employment. Weiss:Merck: Other: Speaker; Bayer: Other: speaker; Genentech: Other: Speaker; NeoGenomics: Current Employment. Brown:NeoGenomics,Inc.: Current Employment. Agersborg:NeoGenomics: Current Employment.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal